Phonon Analysis¶
PDGui performs an analysis of a phonon mode in terms of the percentage of molecular centre-of-mass and rigid body rotational motion in each mode. Using the results of the analysis it is possible to differentiate between inter- and intra-molecular phonon modes (see for instance Jepsen et al [55]). In addition, for those systems with more than one formula unit in the unit-cell it is possible to break down the contribution of each molecule to a particular phonon mode.
Molecular Systems¶
It is common in molecular calculations of vibrational properties to construct a force constant matrix which enforces the requirements of zero energy change for centre-of-mass motion and rigid-body rotation. This can be achieved by using projection operators to transform the second derivative matrix to a set of coordinates that no longer include centre-of-mass motion or rigid-body rotation.
Defining a projection operator as;
where \(\fieldbf{V}\) is an orthonormal column vector with length \(3N\) (\(N\) is the number of atoms), the centre-of-mass motion can be described in Cartesian space as all atoms moving along the x, y or z axis with the same displacement. So considering the projection of the centre-of-mass motion along the x-axis, for each atom \(a\) we can write;
For the projection operator which will project out all components of translation along the x-axis for every atom in the molecule we have;
There are two other projection operators describing translation along the y- and z-axis.
In a similar fashion, it is possible to describe an infinitesimal molecular rotation using a vector V and therefore constructing a projection operator to remove rigid-body rotation. If the coordinates of atom \(a\) relative to the centre-of-mass of the molecule are \(x, y \text{ and } z\), the component of the projection vector, \(\tensorbf{V}\) representing rotation about the x-axis in the yz-plane is;
In a similar fashion rotations about the y- and z-axis are respectively;
and
In a similar fashion to Equation (77) the complete projection operators for the 3 rotational modes can be assembled (using rotation about the x-axis as an example)
In practice, the projection operators will be defined using mass-weighted cartesian coordinates as this simplifies the expressions used later. As an example the transformation to mass-weighted coordinates for atom \(a\) translating along the x-axis is;
Periodic Systems¶
The invariants of the energy in a periodic system are the three translational modes corresponding to motion of all the atoms in the same direction. The rotation of an infinite lattice is not invariant. However, it is proposed to construct projection operators for each molecule in the unit-cell, which will project out the motion of each molecular translational and rotational degree of freedom. Such projection operators will be used to separate the external from the internal modes and there will therefore be a projection operator for each external mode in the system.
Hug and Haesler have shown [56] that the vibrational kinetic energy can be decomposed into single centre atomic contributions by considering the kinetic energy, \(T_p\), of a normal mode \(Q_p\);
Where the sum is over atoms, \(a\), with mass \(m_a\), \(\fieldbf{\Delta \dot{x}}_{a,p}\) is the time derivative of the Cartesian displacement vector of atom \(a\) along the \(p^{th}\) normal mode and \(\fieldbf{L}_p\) are the components of the orthogonal transformation relating the \(p^{th}\) normal mode and mass-weighted Cartesian displacements. Thus \(\fieldbf{L}\) are the eigenvectors of the mass-weighted second derivative matrix. The atom subscript is used to indicate that only those atomic coordinates involving atom \(a\) are being considered. Derivation of this expression has made use of the following relationship between the mass-weighted normal mode;
As proposed by Hug and Haesler [56], since the kinetic energy can be expressed as atomic contributions, consideration of the Virial theorem indicates that this is true also for the potential energy. Using mass-weighted Cartesian coordinates for convenience, an analysis of the phonon modes may therefore be constructed in the following way. A given phonon mode, \(p\), will have its total kinetic energy partitioned between the molecules in the unit-cell according to;
To calculate the contribution a particular mode has to the centre-of-mass kinetic energy we use the projection operators given in Equation (77) (now in mass-weighted Cartesian coordinates) where only the atoms in that molecule are used to construct the operator;
This approach is used to define 6 projection operators for each molecule. The centre-of-mass energy contained in the \(p_{th}\) mode can be written as;
and the rigid-body rotational energy associated with the mode is;
The molecular vibrational contribution, which can be used to classify the internal modes of the system, can be obtained by subtracting the centre-of-mass and rigid-body rotational; energies from the total;
Since all of the energy terms depend in the same way on \(\dot{Q}_p\), it is not required in the calculation of the relative contributions to the energy coming from the external (molecular centre-of-mass and rigid-body rotation) and the internal modes (vibrational contributions)
Implementation¶
In order to calculate the relative internal and external contributions to each phonon mode it is first necessary to identify the molecules in the crystal. In many cases, the unit-cell is packed with atoms in such a way that the cell is filled, rather than in a way reflecting the bondedness of the molecules. The program first replicates the atoms in all cells neighbouring the central unit-cell. Within this supercell the bonds are determined by calculating the distances between all atoms in the supercell. In practice, an order N method is used whereby instead of searching all the supercell only the space around each atom is searched for potentially bonded partners. The criterion of the presence of a bond between atoms \(i\) and \(j\) is given by the requirement that the distance between the atoms \(r{ij}\) is less than the bonding requirement;
Here \(radius_i\) is the covalent radius of atom \(i\), \(scale\) and \(toler\) are factors that can be altered when running the program.
Once the bonding in the supercell has been determined the program starts with the first atom in the central cell and determines all atoms which are connected to the molecule that it is in. If there are any remaining atoms in the central cell which are not bonded yet then further molecules are added until all atoms in the central cell have been allocated to a molecule.
Finally a new cell with the same dimensions as the original is constructed. Where necessary each molecule is shifted into the cell so that the molecule’s centre-of-mass lies within it.
The projection operators, Equation (87), can now be constructed and the relative energies calculated using Equations (88), (89) and (90).
Examples¶
Isoleucine¶
The example of a calculation on isoleucine using Castep is available in the Examples/Castep/Isoleucine directory of the distribution. The analysis was performed using PDGui. The program first finds the four molecules of isoleucine in the unit-cell of the crystal structure. The ordering of the atoms at this point is different to the ordering in the original cell and in the QM calculation. The visualiser shows the atoms in the new positions. These will not be the same as in the original calculation. The molecules and their centres of mass are summarised in Table 6.
Molecule |
Mass |
CMa |
CMb |
CMc |
|---|---|---|---|---|
0 |
131.172 |
0.293600 |
0.051683 |
0.304906 |
1 |
131.172 |
0.706400 |
0.551683 |
0.695094 |
2 |
131.172 |
0.793231 |
0.588919 |
0.287125 |
3 |
131.172 |
0.206769 |
0.088919 |
0.71287 5 |
Table 7 shows the results of the analysis of the phonon modes with frequencies below 100 cm-1. The first 3 modes should have zero frequency as they are the translationally invariant modes of the lattice. The negative number shown here actually indicates that the mode has an imaginary frequency. As can be seen under the column %mol-cme they are almost completely associated with molecular centre-of-mass motion. This is true also of the lowest non-zero frequency at 32.81 cm-1 and to some extent of the next mode at 39.73 cm-1. However, for this latter mode there is a considerable contribution from rigid-body rotational motion (see the %mol-rot column). As the frequency of the phonon mode increases the contribution from rigid-body motion generally decreases and the contribution from vibrational modes (see the %vib column) increases. The total contribution from molecular motion is summarised in the last four columns.
Freq(cm-1) |
%cm |
%rot |
%vib |
%mol-0 |
%mol-1 |
%mol-2 |
%mol-3 |
|---|---|---|---|---|---|---|---|
-0.07 |
99.5 |
0.5 |
0.0 |
26.9 |
26.9 |
23.1 |
23.1 |
-0.06 |
98.6 |
1.1 |
0.3 |
23.6 |
23.6 |
26.4 |
26.4 |
-0.04 |
99.7 |
0.2 |
0.1 |
23.8 |
23.8 |
26.2 |
26.2 |
32.81 |
92.0 |
5.9 |
2.1 |
26.8 |
26.8 |
23.2 |
23.2 |
39.73 |
77.9 |
19.6 |
2.5 |
22.1 |
22.1 |
27.9 |
27.9 |
49.42 |
10.4 |
81.9 |
7.7 |
19.0 |
19.0 |
31.0 |
31.0 |
52.12 |
80.7 |
17.3 |
2.0 |
17.6 |
17.6 |
32.4 |
32.4 |
54.15 |
73.4 |
19.0 |
7.5 |
21.2 |
21.2 |
28.8 |
28.8 |
59.59 |
37.7 |
56.6 |
5.7 |
23.9 |
23.9 |
26.1 |
26.1 |
62.05 |
77.3 |
17.2 |
5.5 |
23.4 |
23.4 |
26.6 |
26.6 |
69.91 |
9.6 |
69.4 |
21.0 |
17.6 |
17.6 |
32.4 |
32.4 |
73.19 |
7.3 |
61.1 |
31.6 |
6.6 |
6.6 |
43.4 |
43.4 |
76.00 |
13.6 |
61.7 |
24.7 |
31.6 |
31.6 |
18.4 |
18.4 |
76.25 |
3.2 |
48.1 |
48.7 |
10.6 |
10.6 |
39.4 |
39.4 |
80.49 |
31.5 |
43.8 |
24.7 |
19.3 |
19.3 |
30.7 |
30.7 |
87.54 |
37.9 |
35.4 |
26.7 |
26.8 |
26.8 |
23.2 |
23.2 |
89.31 |
12.2 |
58.1 |
29.6 |
27.5 |
27.5 |
22.5 |
22.5 |
90.49 |
16.9 |
53.9 |
29.2 |
22.4 |
22.4 |
27.6 |
27.6 |
94.51 |
49.1 |
36.9 |
14.0 |
39.4 |
39.4 |
10.6 |
10.6 |
94.79 |
47.0 |
16.9 |
36.1 |
23.0 |
23.0 |
27.0 |
27.0 |
BaTiO3¶
The example for BaTiO3 can be found in Example/AbInit/BaTiO3 in the main directory of the PDielec distribution. The results of the Abinit calculation can be analysed using PDGui. For this example, the default covalent radius of the Barium atoms has been changed to 0.3 Å, so this atom will be treated as though it is not bonded to anything else in the cell. This results in 2 ‘molecules’ being found in the cell; a TiO3 moiety and the Ba2+ ion.
The results of the analysis are shown in Table 8 below. In this example, the lowest 3 modes have been projected so there is no problem with translational invariance. They are purely centre-of-mass modes. The energy in these three modes seems to be mainly in molecule 1, which is the Ba2+ ion. The modes at 197.73 cm-1 also have a large centre-of-mass component, this time mainly coming from molecule 0, which is the TiO3 moiety. Above 200 cm-1 there is a much smaller centre-of-mass contribution to the energy and all the modes are dominated by the TiO3 group.
Freq(cm-1) |
%cm |
%rot |
%vib |
%mol-0 |
%mol-1 |
|---|---|---|---|---|---|
0.00 |
100.0 |
0.0 |
0.0 |
41.1 |
58.9 |
0.00 |
100.0 |
0.0 |
0.0 |
41.1 |
58.9 |
0.00 |
100.0 |
0.0 |
0.0 |
41.1 |
58.9 |
197.73 |
72.9 |
6.7 |
20.4 |
70.0 |
30.0 |
197.73 |
72.9 |
6.7 |
20.4 |
70.0 |
30.0 |
197.74 |
72.9 |
6.8 |
20.4 |
70.0 |
30.0 |
269.34 |
27.1 |
20.3 |
52.7 |
88.9 |
11.1 |
269.34 |
27.0 |
20.3 |
52.7 |
88.9 |
11.1 |
269.34 |
27.0 |
20.3 |
52.7 |
88.9 |
11.1 |
270.59 |
0.0 |
55.6 |
44.4 |
100.0 |
0.0 |
270.59 |
0.0 |
55.6 |
44.4 |
100.0 |
0.0 |
270.59 |
0.0 |
55.6 |
44.4 |
100.0 |
0.0 |
493.61 |
0.1 |
17.4 |
82.5 |
100.0 |
0.0 |
493.61 |
0.1 |
17.4 |
82.5 |
100.0 |
0.0 |
493.61 |
0.1 |
17.4 |
82.5 |
100.0 |
0.0 |
The visualisation of the internal and external contributions to the phonon modes is shown in the Fig. 17
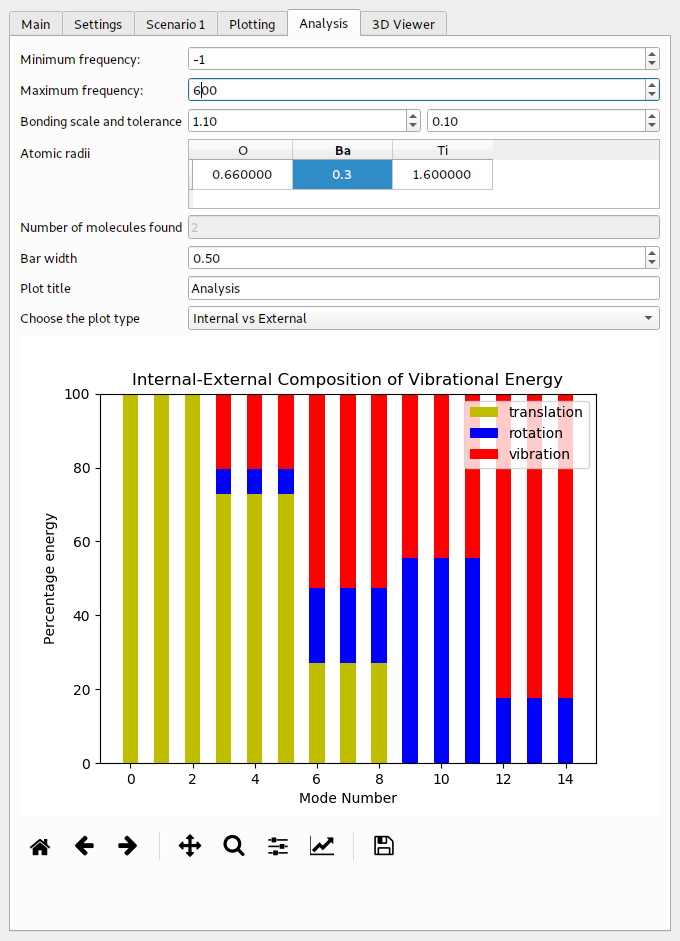
Fig. 17 Internal and external contributions to the phonon modes of BaTiO3¶