PDGui¶
PDGui provides a graphical front end to the capabilities of PDielec. PDGui can be run from the command line without any parameters or followed by the file to be read in. PDGui makes a best guess at the DFT/MM program used to create the output. It can also be used as a command line tool without having to use the GUI, this needs a script file to be available.
For example;
pdgui
or
pdgui OUTCAR # A Vasp calculation, only OUTCAR is parsed for information
pdgui vasprun.xml # A Vasp calculation, only vasprun.xml is parsed for information
pdgui LEUCINE_FREQUENCY_PBED3_631Gdp_FULLOPTIMIZATON.out # A Crystal calculation
pdgui run1.dynG # A QE calculation, pwscf.xml is also read
pdgui run1.log # A QE calculation, run1.dynG is also read
pdgui run2.abo # An Abinit calculation
pdgui run3.gout # An Gulp calculation
pdgui phonopy.yaml # A Phonopy calculation
pdgui control.in # An FHI-Aims calculation
Sometimes pdgui is unable to unambiguously determind which DFT program has been read. This is particularly problematic for phonopy calculations. It is possible to specifiy the DFT program as in the examples below;
pdgui -program aims geometry.in # An FHI-Aims calculation
pdgui -program vasp OUTCAR # A Vasp calculation, only OUTCAR is parsed for information
pdgui -program phonopy phonopy.yaml # A Phonopy calculation
If running a script that contains program and output file information, then:
pdgui -script script.py
If a spreadsheet of results is required the name of the spreadsheet can be provided on the command line.
pdgui OUTCAR -spreadsheet results.xlsx
The spreadsheet name must have an .xlsx extension. To use as a command line tool an example would be;
pdgui OUTCAR -spreadsheet results.xlsx -script script.py -nosplash -exit
Several command line options may be useful in running the package.
Command line option |
Description |
|---|---|
-script filename |
Specify a Python script to run |
-scenario type |
Changes the initial scenario type, type can be powder or crystal, the default is powder |
-spreadsheet file |
An alternative way of defining the spreadsheet |
-program name |
An alternative way of defining the program used to create the output |
-version |
Give information of the version no. |
-debug |
Debugging mode, only for developers |
-exit |
Exit the program after running the script |
-nosplash |
Do not show the splash screen |
-cpus 2 |
Sets the number of cpus or threads to 2 |
-threads |
Using threading, instead of multiprocessing |
-h |
Print out help information |
After running the program the user sees a notebook interface with seven tabs.
Main Tab allows the user to specify the program and filename that are to be analysed. It is also possible to specify here the spreadsheet name to which results are saved.
Settings Tab is used for changing the settings within the package
Scenario Tab can be a powder scenario or a single-crystal scenario. In the case of a powder scenario, the tab specifies the material properties used to define the effective medium from which the absorption characteristics are calculated. In the case of a single crystal, the orientation of the crystal and the angle of incidence of the radiation can be defined.
Plotting Tab shows the results of all the scenario tabs in one place.
Analysis Tab shows the decomposition of the normal modes into molecular components.
Visualisation Tab displays the normal modes as either arrows showing the atomic displacement or as an animation.
Fitter Tab allows a user to display an experimental spectrum and compare it with the calculated spectrum.
Main Tab¶
The Main Tab is used to pick the MM/QM package and the output file that will be analysed.
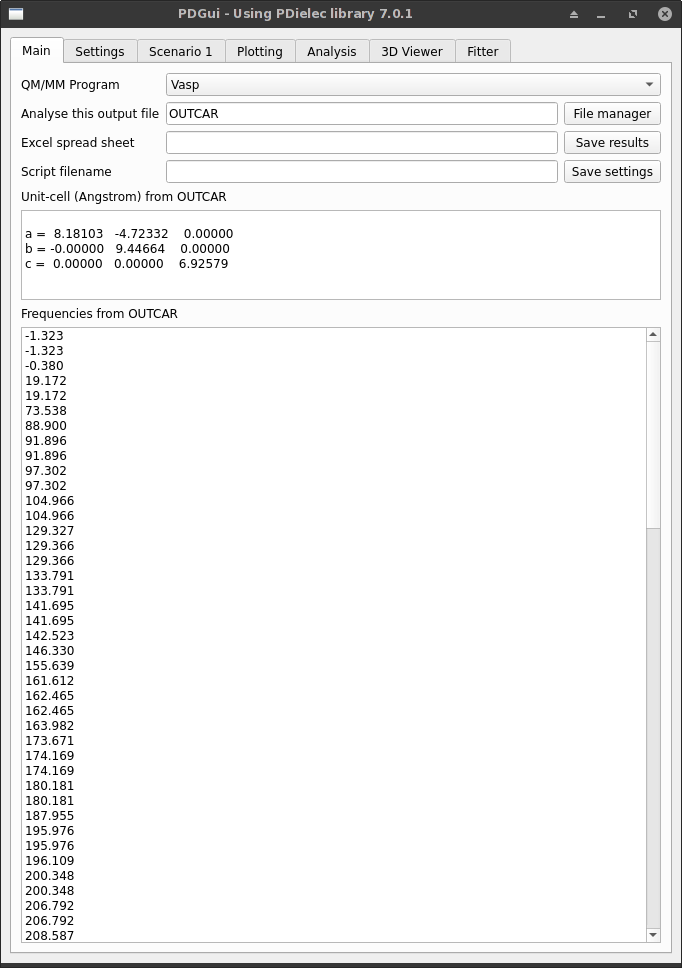
Fig. 1 The Main Tab¶
The QM/MM program can be chosen from the dropdown list. The current list of packages supported is Abinit, Castep, Crystal, Experiment, Gulp, FHI-Aims, Phonopy, Quantum Espresso, Vasp, and PDGui. The output file name can be provided in the text box below it, or it can be specified using the File manager button. In addition to input files from QM/MM programs, the program can read a Python script files written by PDGui. This should have a ‘.py’ extension. The Experimental file format allows the user to provide their specification of a dielectric medium, where a code other than that catered for by the program has been used. The file format is described in full in the section: Experimental File Format The PDGui option allows the user to select a scripting file from the interface.
If an Excel spreadsheet file name is given, it must have the extensions .xlsx. The spreadsheet is written when the Save results button is pressed.
If the current program settings need to be stored they can be stored as a Python script file by pressing the Save settings button. Any settings saved in this way can be re-instated by reading the script using the File manager and selecting the PDGui (*.py) option in the file types selection.
Once the file has been specified and read, the unit cell is shown and the frequencies found in the calculation file are reported in the output text boxes.
Settings Tab¶
The Settings Tab controls the calculation of the frequencies and their intensities.
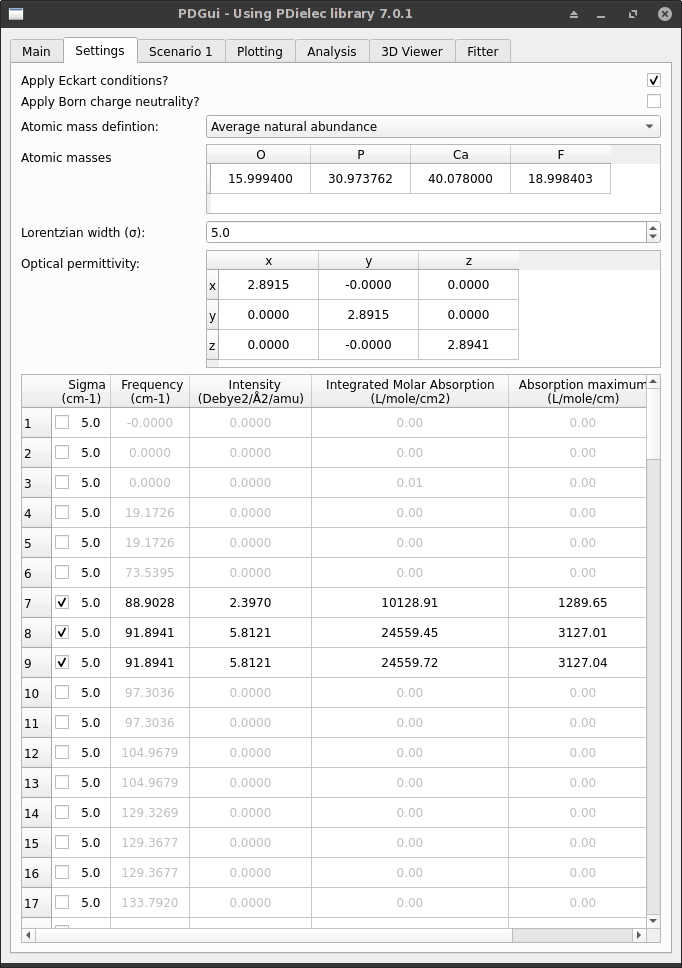
Fig. 2 The Settings Tab¶
The Eckart conditions control whether 3 modes at zero frequency due to translational invariance are projected out of the dynamical matrix. By default, the Eckart conditions are applied and the 3 modes are projected out.
There is an option to set the sum of the Born charge matrices to zero. By default, this is not applied.
- The atomic masses can be specified in a variety of ways. They can be chosen from;
The average natural abundance
The masses used by the QM/MM program
The isotopic mass of the most abundant isotope
If necessary the mass of each element can be edited separately by clicking on the mass concerned and entering a new number.
The width (\(\sigma\)) of all the absorptions can be set using the Lorentzian width (sigma) spin box.
Finally, the optical permittivity at zero frequency is given in the Optical permittivity table. In some cases, it is necessary to enter the optical permittivity by hand. This can be done by clicking each element in the table that needs changing and typing the new matrix element.
When a new output file is read the atomic masses and the optical permittivity matrix are reset.
The output table at the bottom of the tab shows the calculated frequencies and their intensities. Transitions that do not contribute to the Infrared absorption are greyed out. These transitions will not be used in later calculations. From this table it is possible to remove or add transitions to the later calculations and it is also possible to change the width of individual transitions.
Scenario Tabs¶
There are two sorts of scenario tab, one for powder calculations and one for single-crystal calculations. By default, it is assumed that powder calculations are being performed. There is a button (Switch to crystal scenario or Switch to powder scenario) at the bottom of the tabs which allows the user to switch between the two.
Scenarios can be added or removed using the push buttons at the bottom of each Scenario Tab. When a new scenario is created the settings are copied from the current scenario.
There can be more than one Scenario Tab. Each one specifies a particular material, method, or particle shape which will be used for the calculation of the effective medium or single crystal. The results of the calculations will be shown in the Plotting Tab.
Powder Scenarios¶
For each powder scenario it is assumed that the size of the particle embedded in the matrix is small compared with the wavelength of light.
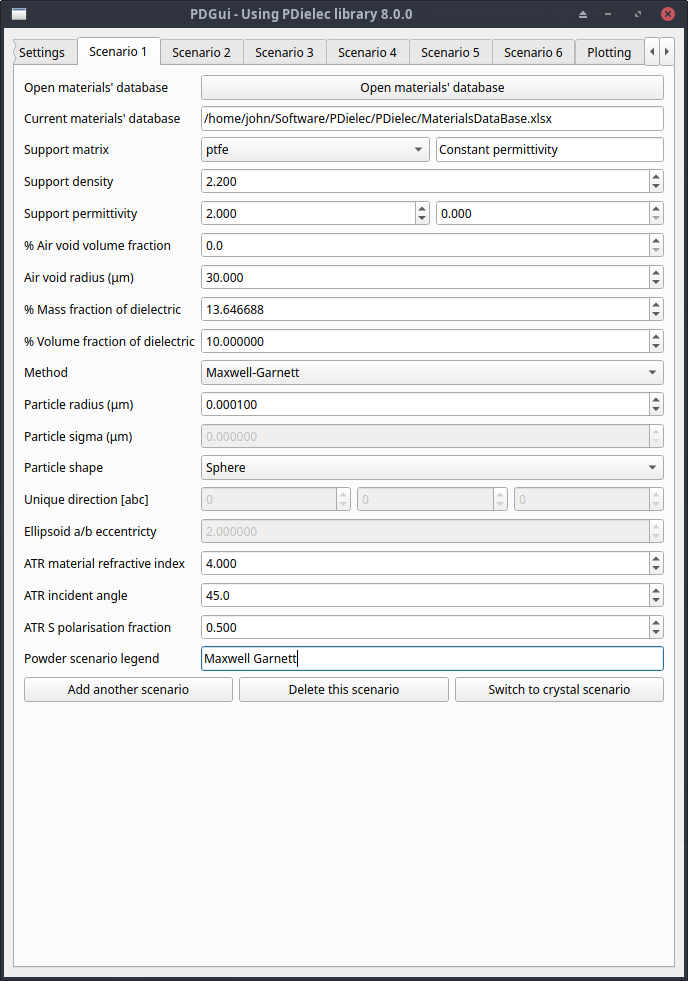
Fig. 3 The Scenario Tab for a Powder¶
A database of material properties is opened at the start of the program. The default database (PDielec/MaterialsDataBase.xlsx) is opened although other databases can be loaded by pressing the Open materials’ database button. The open database is shown in the line below, followed by a line showing the support matrix material selected along with a brief description of the type of entry for the material.
Materials can have either a constant or frequency-dependent permittivity. In the case of frequency-dependent entries the range of frequencies which the entry covers is displayed. A warning is appropriate here as all of the frequency-dependent materials have been extrapolated to be valid at 0 \(cm^{-1}\) and above. The extrapolated values are indicated in the database itself.
The support matrix into which the active dielectric material is dispersed can be selected from the Support matrix drop-down menu. The selected supporting material will change the density and permittivity shown in the respective text boxes. For frequency-dependent entries the value of the permittivity at 0 \(cm^{-1}\) is used. The user can edit these values independently if necessary. But if this is done the program assumes that the changes result in a frequency-independent permittivity. The Support permittivity consists of two inputs for the real and imaginary components of the permittivity. For any Mie calculation the imaginary component is ignored.
The supporting medium may have bubbles of air trapped in the matrix. For the case that polyethylene spheres are used to make the sample, experimental information indicates a relationship between the size of the spheres and the size of the air inclusions [18].
PE Sphere Diameter (μ) |
Porosity (%) |
Void radius (μ) |
|---|---|---|
60 |
24 |
24.0 |
72 |
25 |
28.0 |
360 |
44.5 |
90.0 |
The size of the air inclusions (bubbles) can be specified in the Scenario Tab along with the volume fraction. If a non-zero value for the volume fraction of air voids is given, the scattering due to the air inclusions is calculated before the effective medium calculation is performed. The forward and backward scattering amplitudes for a single scatterer are calculated using the Mie method and then used to calculate the scattering using the Waterman-Truell approximation.
The amount of dielectric material can be entered either as a mass fraction (in percent) or as a volume fraction (in percent). If the matrix support density is changed the calculated mass fraction will be updated. It is assumed that the volume fraction has precedence. If an air void volume fraction is supplied then this is taken account of in the calculation of the volume and mass fractions.
The calculation of the effective medium can be performed using a variety of methods which can be chosen from the Method drop-down menu. If the Mie method is chosen the user can enter the particles’ radius (μ). Particle sigma specifies the width of a log-normal distribution. If the width is 0.0 no sampling of the distribution is performed.
For effective medium theories, the particle shape can be specified using the Shape pull-down menu. Possible shapes are Sphere, Needle, Plate and Ellipsoid. For the cases of Needle and Ellipsoid the unique direction is specified by a direction [abc] in lattice units. In the case of Plate the unique direction is specified as the normal to a plane (hkl) in recipricol lattice units. If an Ellipsoid shape is used the eccentricity factor can be specified in the Ellipsoid a/b eccentricity text box.
The plot in the Plotting Tab has a legend and the description used in the legend for each scenario can be set by filling out the text box associated with the Scenario legend.
Single-Crystal Scenarios¶
As in the powder scenario, a single-crystal scenario has an option to open a new database of materials. By default, the program opens the default database distributed with the program.
The mode of calculation is determined by the Single crystal methodology option. Possible options are; Transfer matrix and Scattering matrix. Details of the theory underlying each method are given in the theory section: Theory for Single Crystal Optics. The transfer matrix is faster and all the available methods for treating incoherence are available. The scattering matrix method is slower and the option to treat incoherence by using intensities is not available, but it is much more stable when treating thick films. Single-crystal films are defined by a surface determined by the (hkl) settings.
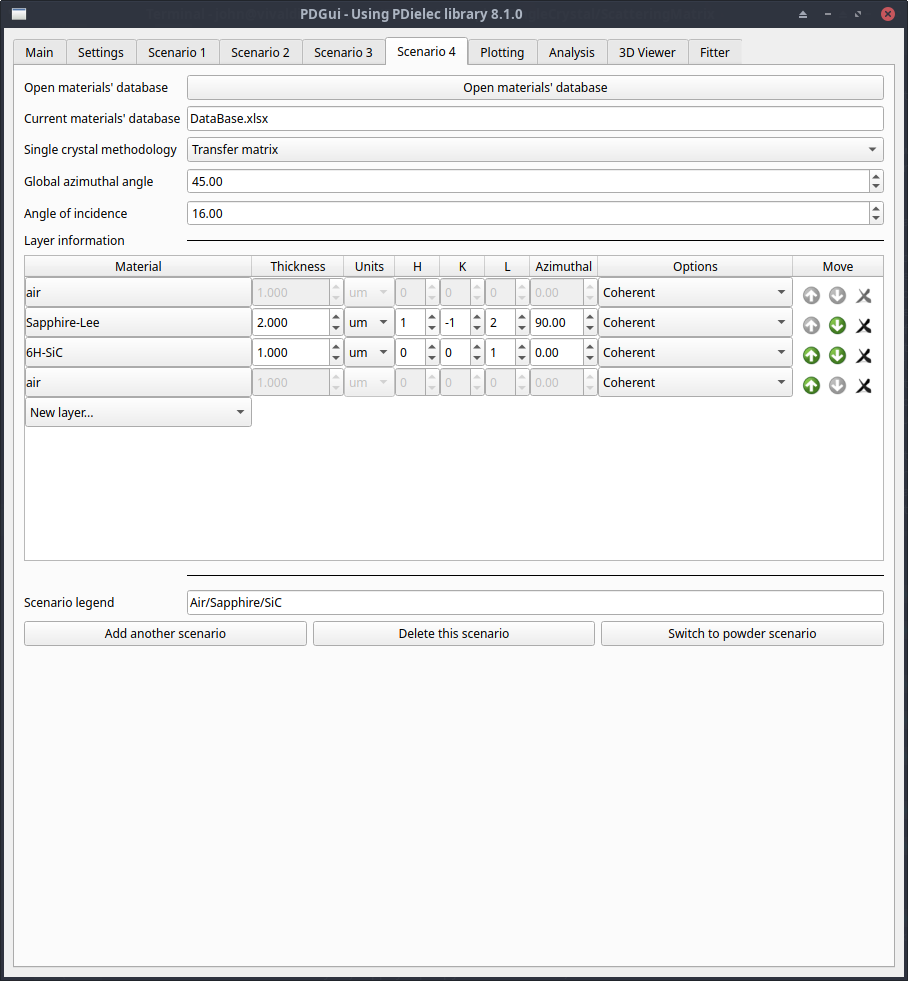
Fig. 4 The Scenario Tab for a Single Crystal¶
The method of calculation is selected at the top of the tab, followed by the global azimuthal angle and the angle of incidence. The global azimuthal angle controls the rotation of all the layers around the laboratory Z-axis, although each layer also has its own azimuthal angle as will be described later.
The layer information is provided as a spread-sheet of materials, with the specification of the thickness of the layer, the (hkl) parameters of the surface, the azimuthal angle of the layer and an option to treat the layer coherently or incoherently. The material at the top of the list is the superstrate, which should be isotropic and non-absorbing. It is treated as a semi-infinite material. The material at the bottom of the list is the substrate, which is also semi-infinite. Although, if only reflectance information is needed, this can be an anisotropic, absorbing material. The layer in-between is initially set to the DFT dielectric material. New layers can be added by pressing the New layer… button and choosing from what is available in the database. The position of the layers can be adjusted by pressing the up and down buttons and a layer can be removed by pressing the delete button. There are options to treat the layer, coherently or incoherently available from the Options pull down.
Pressing the button which shows the name of the material, brings up the layer editor window for that material, which is described in more detail below.
Finally, there is a legend that can be provided for the Plotting Tab.
The Layer Editor¶
To see more details of a crystalline layer in the system press the Material button of the material of interest. This brings up a new window, which shows more information about the layer. A useful feature of the layer editor is that it shows the connection between the crystal and laboratory coordinate systems. The laboratory axes are defined with Z- being normal to the surface (hkl), and the incident and reflected beams lie in the XZ- plane, which means the laboratory Y- axis is perpendicular to the XZ- plane. The incident radiation is assumed to be at an angle \(\theta\) to the normal. p-polarised light lies in the XZ- plane and s-polarised light is parallel with the Y-axis. The azimuthal angle \(\phi\) defines the rotation of the crystal around the Z-axis. To help understand the relationship between the crystal and laboratory coordinates the program outputs the direction of the lattice vectors in terms of the laboratory X-, Y- and Z- coordinates. A more complete description of the relationship between the coordinate systems is given in Crystal & Laboratory Coordinate Frames
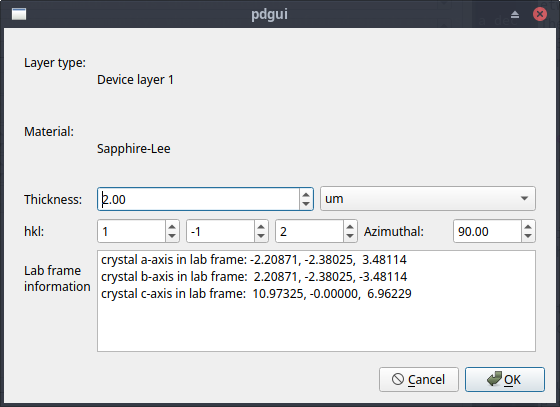
Fig. 5 The layer editor¶
Plotting Tab¶
The Plotting Tab controls and plots the absorption and permittivity as a function of frequency calculated for each scenario present in the pdgui notebook. The plots available depend on the availability of powder and single-crystal scenarios.
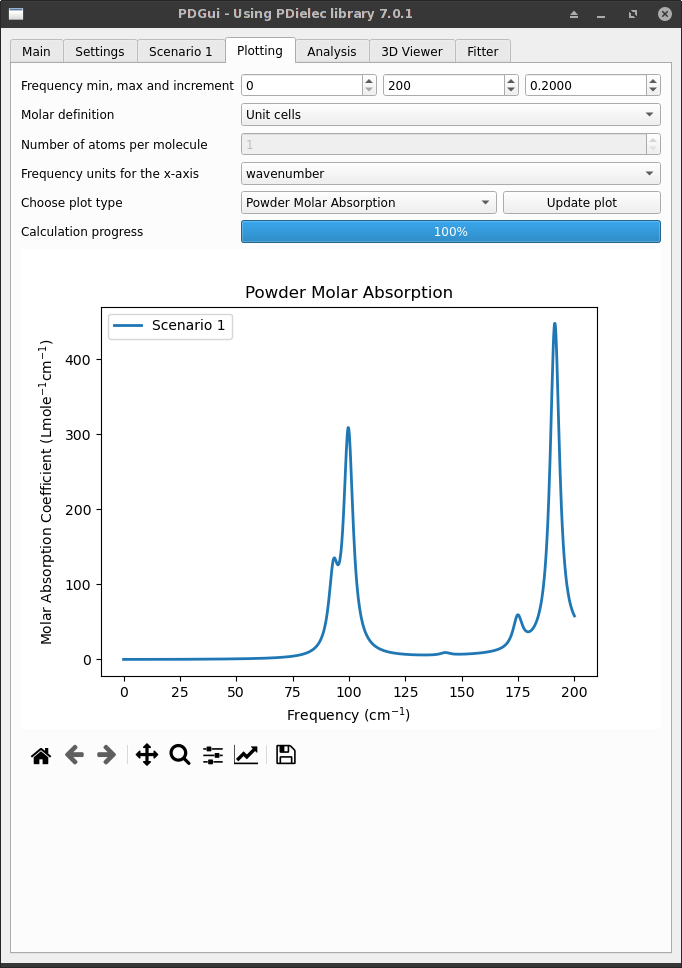
Fig. 6 The Plotting Tab Showing Powder Absorption¶
The minimum and maximum frequencies can be specified along with the frequency increment. An effective medium theory calculation will be performed for each scenario at every frequency between the minimum and maximum frequencies at the interval specified.
By default, the calculation uses moles of unit cells for the molar absorption coefficient. This can be altered using the the Molar definition pull-down menu. Options include Unit cells, Atoms and Molecules. In the case of Molecules, it is necessary to supply the number of atoms in a formula unit of the compound in the Number of atoms per molecule text box.
The title of the plot can be supplied by entering it into the Plot title text box and the frequency units used for the plot can also be changed from wavenumber to THz.
For powder scenarios, the molar absorption, the absorption, and the real or imaginary permittivity can be plotted. Once a plot has been requested the calculation progress is shown in the progress bar. Some settings can be changed without the whole plot being recalculated.
For single-crystal plots, the crystal reflectance and transmittance for s and p polarised radiation can be plotted along with absorptance which is defined in terms of the reflectance (R) and the transmittance (T) as \(1-R-T\). For thick slabs, only the reflactance is of any relevance. For thin films, absorptance is useful as it removes some of the oscillations that occur in the transmittance or reflectance due to the film thickness.
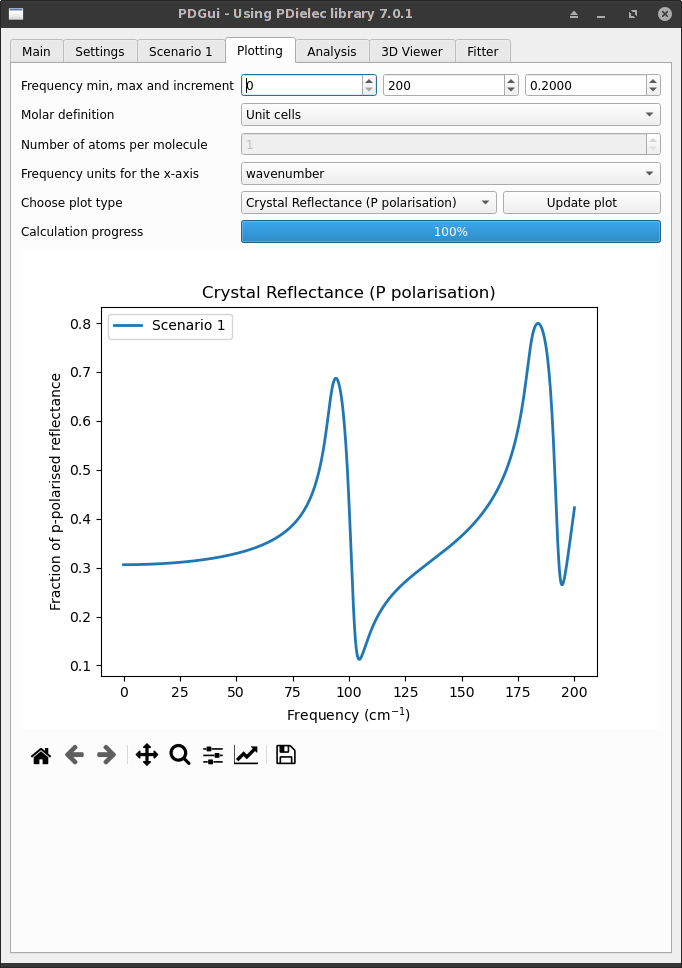
Fig. 7 The Plotting Tab showing Single-Crystal results¶
The title of the plot can be supplied by entering it into the Plot title text box and the frequency units used for the plot can also be changed other units including wavenumber, THz or a wavelength such as cm, mm or um. A default plot title is generated giving information about the type of plot being viewed.
Analysis Tab¶
The Analysis Tab shows a breakdown of the phonon modes into molecular components or internal and external modes. The molecular structure of the unit cell is determined by the covalent radii of the atoms. These can be specified individually if needed. The Analysis Tab shows the number of molecules that have been found in the analysis.
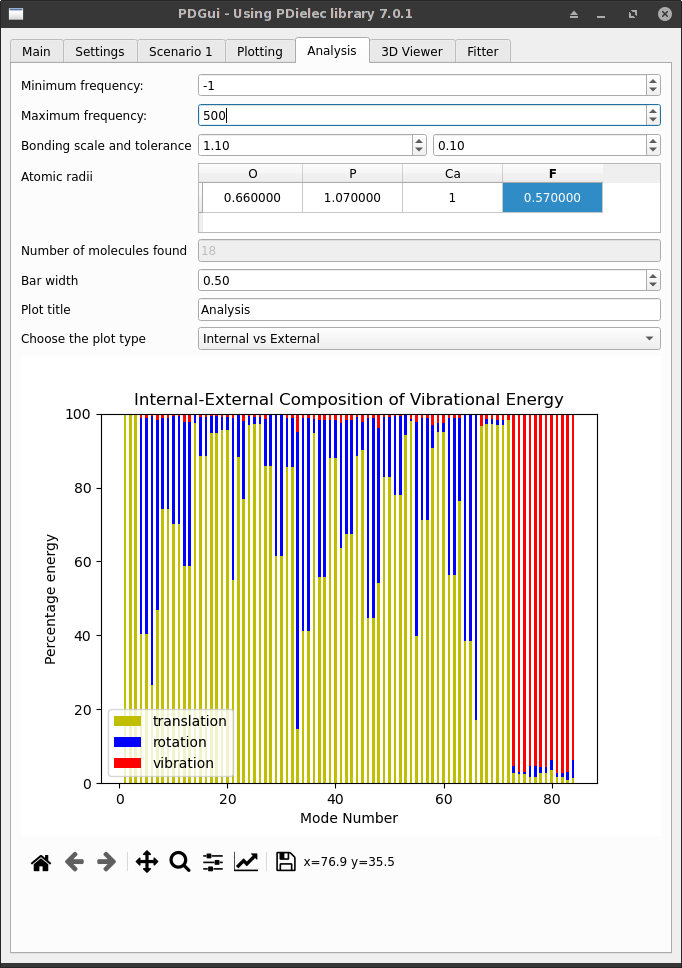
Fig. 8 The Analysis Tab¶
The bar graph shows a breakdown of each normal mode in the chosen frequency range into either internal and external contributions or into molecular components. The atom sizes and the molecular composition of the unit cell are displayed in the 3D Viewer Tab
3D Viewer Tab¶
The 3D Viewer Tab shows the unit cell of the system using the molecular information and atomic sizes from the Analysis Tab.
The atomic displacement of each phonon can either be shown as arrows or as an animation. The views and animations can be recorded in .png and .mp4 files respectively. If a .gif file is specified the animation is recorded but with reduced numbers of colours. A series of CIF files can also be generated which follows the motion of the mode being studied.
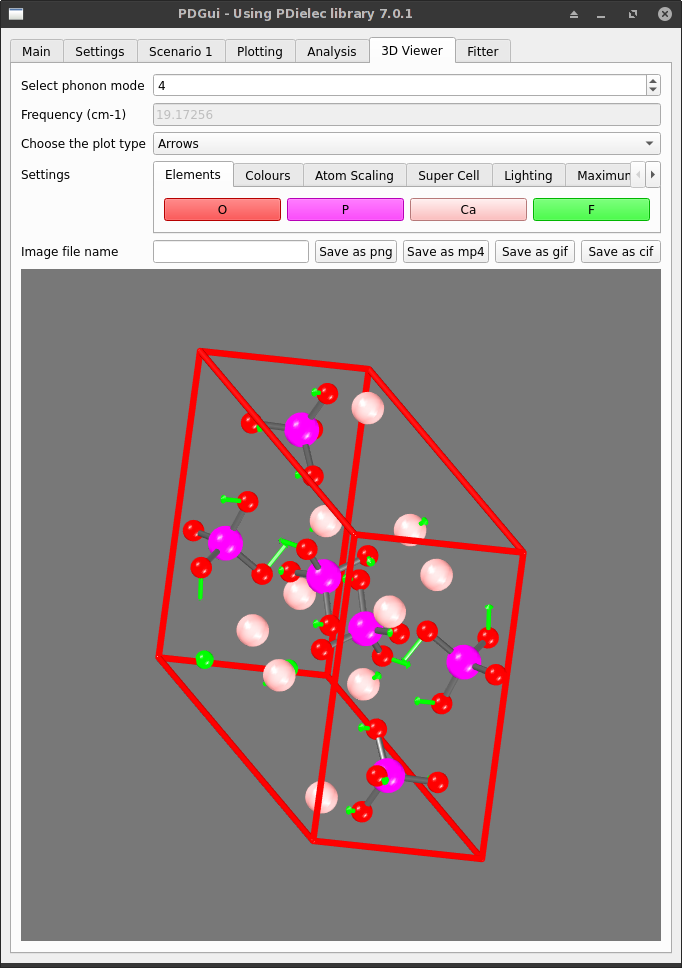
Fig. 9 The 3D Viewer Tab¶
As well as being able to change the phonon mode being analysed. The colours and many settings in the visualiser can be adjusted from the settings tab. The unit cell can be shown as a supercell by altering the a, b, c parameters in the settings tab. Other parameters affecting the display can also be modified.
Fitter Tab¶
The Fitter Tab imports an experimental spectrum. The spectrum is stored in an Excel spreadsheet. The spreadsheet should contain a single sheet with two columns. The first column should be the frequency in cm-1 and the second should be the measured signal. The signal can be molar absorption, absorption, real permittivity, imaginary permittivity, ATR absorbance or any of the single-crystal properties. Once imported the experimental spectrum can be compared with the calculated spectrum. The tab shows the frequencies contributing to the spectrum and allows the Lorentzian widths of the transitions to be altered. The frequency range used for the display is the same as that used in the plotting tab. Indeed the calculated signal used for fitting is the same as that being shown in the plotting tab. At the top of the tab are some settings in a tabbed notebook. The most important is the name of the Excel file containing the experimental spectrum. In addition, there are options to change the Plot type, include frequency scaling in any fitting, set the frequency scaling factor, set the number of iterations to be used when fitting, choose whether the plot should use independent y-axes for the calculated and experimental spectra, set the method used to do the fitting and finally specify the spectral difference threshold. The spectrum is shown at the bottom of the tab and is recalculated when the Replot or Replot with frequency shift buttons are pressed. The data type stored in the experimental spreadsheet is defined by the Plot and data type setting. One of the settings options is the ability to remove a baseline from the experimental spectrum. If baseline removal is selected a Hodrick-Prescott filter is used. The value of the filter parameter is determined by the value in the HP filter lambda tab. The actual value of lambda used in the filter is the value given in the tab raised by the power of 10.
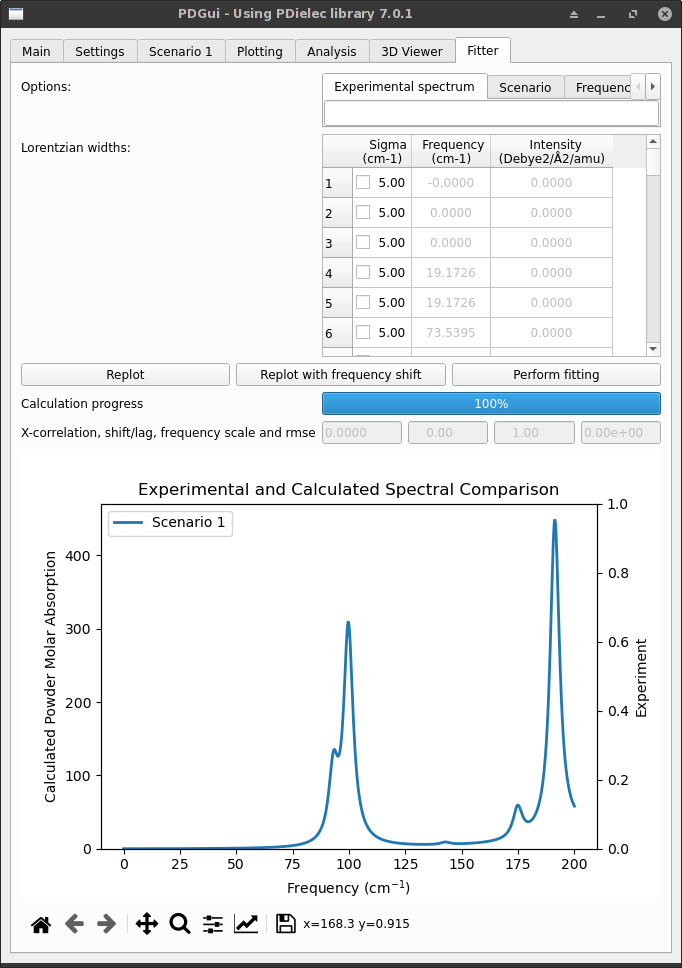
Fig. 10 The Fitter Tab¶
After a replot the cross-correlation coefficient, current frequency scaling factor, the frequency shift needed to maximise that cross-correlation coefficient, and the root mean squared error between the calculated and experimental spectra are shown. It is possible to apply a frequency scaling to the calculated spectrum. The Replot with frequency shift shows the calculated spectrum with the frequencies shifted by the shift calculated to maximise the cross-correlation coefficient. Care must be taken in comparing the calculated and experimental spectra as different y-axes are used for each.
It is also possible to automatically adjust the Lorentzian width factors with the Perform fitting button. However, experience with this option shows that it may be better to adjust the peak heights manually by altering the sigma values. At the moment only 20 iterations are performed for each press of the button. If requested the frequency scaling factor can be adjusted too.
Two algorithms for performing the fitting are supported. Either the cross-correlation coefficient can be maximised or the root mean squared error between the spectra can be minimised. In the latter case the error is only calculated for signal strengths above the spectral difference threshold (which defaults to 0.05).
A summary of the optional settings available in the Fitter Tab is shown below;
Fitter options |
Description |
|---|---|
Experimental spectrum |
Provide a file with the experimental data. A return brings up a file manager |
Scenario |
Allows the user to choose which will be used in the fitting |
Frequency scaling factor |
The calculated frequencies are scaled |
No. of iterations |
Number of iterations for automatic fitting |
Independent y-axes |
The calculated spectrum uses the left axis and the experimental the right |
Fitting type |
|
Optimise scaling |
Tick if frequency scaling is to be included in the automatic optimisation |
Spectral difference threshold |
Used if fitting type is minimise spectral difference |
Baseline removal? |
Tick to request that the Hodrick-Prescott (HP) method is used to remove a sloping baseline in the experimental data |
HP filter Lambda |
Provide the lambda value in the HP method |
Scripting¶
Nearly all aspects of the program can be accessed through scripting. The script is provided by the -script command line option or by choosing the PDGui (*.py) file type from the file manager on the Main tab. A very useful feature is the ability to save a script that reflects the current state of the program. This is available from the Main tab.
There are several things to be aware of when using this option to read in settings from one calculation into another. The file will contain parameters that have been calculated whilst reading the input file. For example, the optical permittivities are calculated and stored in this file. If the file is then used on another DFT calculation then the optical permittivity for the settings file will be used and not the one which should have been calculated. This is also true if masses have been defined in one calculation but they may not be appropriate for a different molecule. To use the script for other calculations but with the same scenarios the optical permittivities should be removed and any mass specifications removed.
Parallelisation and threads¶
To improve the performance of the program python parallelisation has been used to parallelise over the frequencies, shapes and methods. By default, this parallelisation spawns additional Python executables, depending on the number of cores available.
In addition to this form of parallelisation the NUMPY library can use multi-threaded BLAS. NUMPY can be compiled with several different BLAS libraries, including; MKL (from Intel), OPENBLAS and ATLAS, or indeed no optimised BLAS library is necessary. To explore the BLAS version compiled with your version of NUMPY please use the test_numpy_1, test_numpy_2 and test_numpy_3 scripts included in the Python/ subdirectory. If you are using MKL or OPENBLAS, the number of threads being used needs to be set with the MKL_NUM_THREADS or OPENBLAS _NUM_THREADS environment variable (sometimes OMP_NUM_THREADS is also used). Use the test routines to determine the optimum for your system. Experience indicates that no performance benefit is obtained with more than two threads.
In the case of Phonopy, the dynamical matrix is read from a yaml file. This is very slow unless the C parser is used. If the C parser is not available a warning is issued and the program reverts to the Python parser.
Finally, the use of non-standard BLAS libraries seems to cause problems with the affinity settings for multiprocessing. It has been noticed that the parallel processes can all end up executing on the same processor. To prevent this, before executing the pdielec and preader scripts it may be necessary to include;
export OPENBLAS_MAIN_FREE=1
For some reason, this also works if the MKL library is being used.
There have been issues in running PDielec on Catalina, MacOS. This appears to be due to the multiprocessing features of Python not working as expected on this operating system. A workaround is to use;
pdgui -threads -cpu 1
This runs the program on a single processor using the threading library for multiprocessing. Sometimes it is more convenient to set these using the environment. Two environment variables are read before any command line options are processed;
export PDIELEC_NUM_PROCESSORS=4
will run pdgui on 4 processors. By default, the number of processors is determined by interrogating the computer. This is achieved, by setting the value of PDIELEC_NUM_PROCESSORS to 0.
export PDIELEC_THREADING=TRUE
will run pdgui using the threading multiprocessing options.
Performance¶
PDielec has been written to make use of multiprocessor computers. On an 8-processor (16 thread) machine using multiprocessing, the speed-up soon slows down after three processors, as there is a lot of the code which is not parallelised . Threading does not appear to be an efficient way of running PDielec and it is only recommended if the default multiprocessing options is failing for some reason. The speedup is calculated by running the benchmark suite (pdmake benchmarks), which consists of 30 different examples of using the code and is therefore representative of the type of calculations performed.
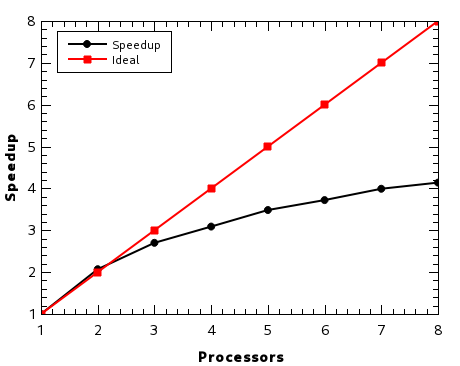
Fig. 11 Speed-up on a 8-processor workstation¶
Using the same suite of benchmarks on an 8-processor Intel core i9-9900K running at 3.6GHz took 92.6s.
On a Windows laptop with an Intel Skylake i7-650, which has 2 processors running at 2.5GHz, the benchmark ran in 432s seconds. On the same machine but running the Linux operating system the benchmark suite took 358s, making the impact of Windows on performance a factor of 1.2 times slower.