Phonopy Examples¶
Phonopy [7] is an open source package for the calculation of phonon’s using the harmonic and quasi-harmonic approximations. Calculations of the phonon spectrum at the \(\Gamma\) point in the Brillouin-zone using Phonopy can be used by PDielec to calculate the absorption spectrum of powdered and crystalline materials. The only additional requirement for performing calculations using PDielec with Phonopy is the provision of \(\epsilon _{\infty}\) and the Born charges. These are also required by Phonopy if the non-analytical correction is necessary. In this case, Phonopy provides a script for several DFT packages which converts the DFT calculation of Born charges to a file BORN, which contains the \(\epsilon _{\infty}\) and the Born charges. For VASP, the script is called phonopy-vasp-born. The BORN file stores only the symmetry unique charges and PDielec provides a command, phonopy-pdielec-born, which reads the BORN file and phonopy.yaml and generates all of the Born charges. The command only uses the Phonopy API, so it is necessary to use it in an environment that has Phonopy installed.
α-Al2O3¶
This example is based on the example/Al2O3 given in the Phonopy distribution. There may be some differences between the present calculations and that in Phonopy’s distribution as the INCAR, KPOINTS and POTCAR files are not present in the distribution.
α-Al2O3 or corundum belongs to the \(R\overline{3}c\) space group. The standard cell has 6 formula units (30 atoms) in the cell, and the primitive cell has 2 formula units (10 atoms).
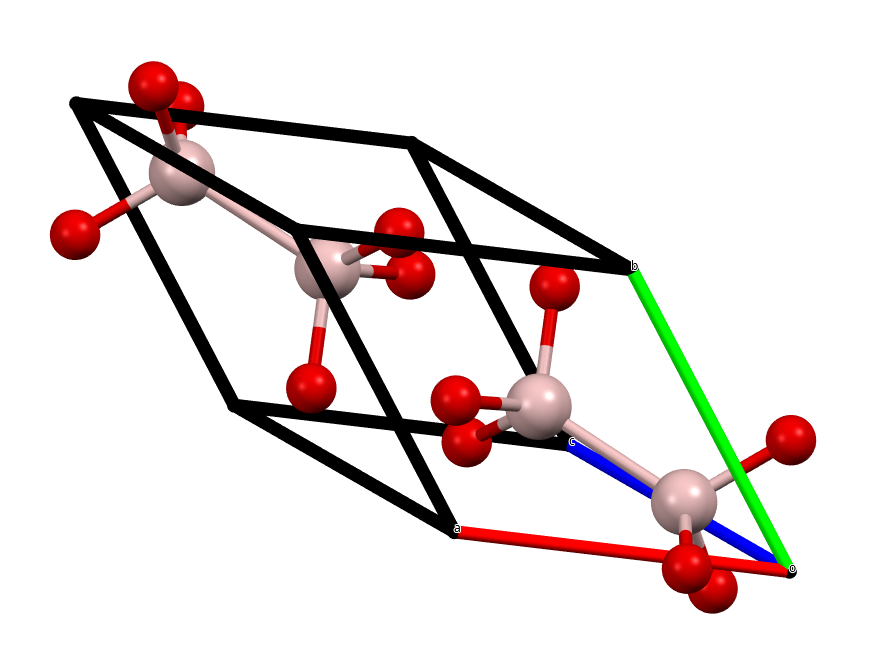
Fig. 65 The primitive cell of Corundum¶
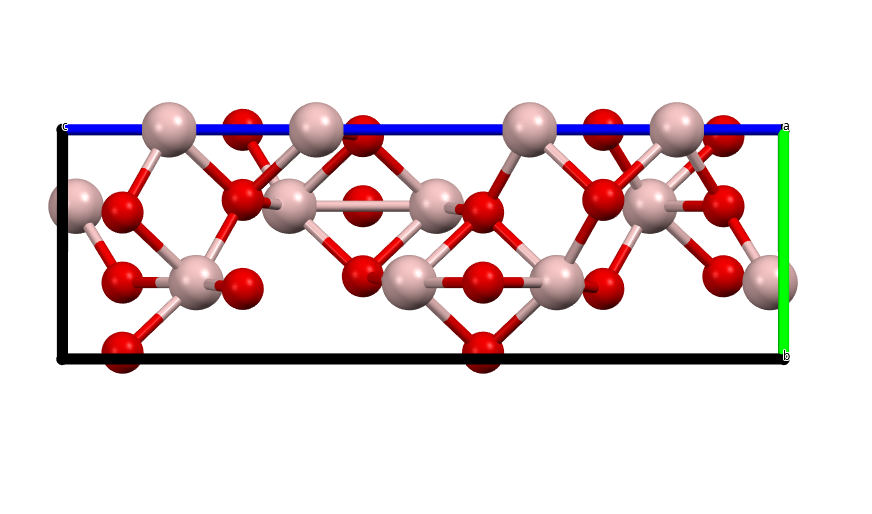
Fig. 66 The standard cell of Corundum¶
In the PDielec’s Examples/Phonopy/Al2O3 directory there are two directories, Standard_cell/ and Primitive_cell, containing results of Phonopy calculations for the standard and primitive cells respectively.
It is assumed that the optimisation of the structure has already been done as indicated below
Geometry Optimisation¶
The INCAR file for used for the optimisation was;
PREC = accurate
ALGO = Normal
ADDGRID = T
NELMIN = 5
IBRION = 2
POTIM = 2
NSW = 50
EDIFF = 1E-8
EDIFFG = -1E-3
ENCUT = 500.00000000
LREAL = F
ISMEAR = 0
SIGMA = 0.01000000
GGA = PS
LMAXMIX = 4
The KPOINTS file is;
Regular k-point mesh
0
Monkhurst-Pack
3 3 1
0 0 0.5
The optimised POSCAR is;
generated by phonopy
1.0000000000000000
4.7742263452989944 0.0000000000000000 -0.0000000000000000
-2.3871131726494972 4.1346012984458653 0.0000000000000000
-0.0000000000000000 0.0000000000000000 13.0113593913272219
Al O
12 18
Direct
0.3333333333333357 0.6666666666666643 0.0187603745624614
0.3333333333333357 0.6666666666666643 0.3145729587708743
0.0000000000000000 0.0000000000000000 0.1479062921042100
0.6666666666666643 0.3333333333333357 0.1854270412291257
0.0000000000000000 0.0000000000000000 0.3520937078957900
0.0000000000000000 0.0000000000000000 0.6479062921042100
0.6666666666666643 0.3333333333333357 0.4812396254375386
0.3333333333333357 0.6666666666666643 0.5187603745624614
0.6666666666666643 0.3333333333333357 0.6854270412291257
0.6666666666666643 0.3333333333333357 0.9812396254375386
0.3333333333333357 0.6666666666666643 0.8145729587708743
0.0000000000000000 0.0000000000000000 0.8520937078957900
0.3058086537880129 0.0000000000000000 0.2500000000000000
0.3608580128786514 0.3333333333333357 0.0833333333333357
0.0000000000000000 0.3058086537880129 0.2500000000000000
0.6666666666666643 0.0275246795453228 0.0833333333333357
0.6941913462119871 0.6941913462119871 0.2500000000000000
0.9724753204546772 0.6391419871213486 0.0833333333333357
0.9724753204546772 0.3333333333333357 0.5833333333333357
0.0275246795453228 0.6666666666666643 0.4166666666666643
0.6666666666666643 0.6391419871213486 0.5833333333333357
0.3333333333333357 0.3608580128786514 0.4166666666666643
0.3608580128786514 0.0275246795453228 0.5833333333333357
0.6391419871213486 0.9724753204546772 0.4166666666666643
0.6391419871213486 0.6666666666666643 0.9166666666666643
0.6941913462119871 0.0000000000000000 0.7500000000000000
0.3333333333333357 0.9724753204546772 0.9166666666666643
0.0000000000000000 0.6941913462119871 0.7500000000000000
0.0275246795453228 0.3608580128786514 0.9166666666666643
0.3058086537880129 0.3058086537880129 0.7500000000000000
Calculation of the Born charges and \(\epsilon _{\infty}\)¶
Phonopy allows for the calculation of the phonons in either the primitive or standard basis. Starting the calculation in the standard cell basis will allow later calculations in the primitive celll basis.
In the Standard_cell/Born directory a calculation of the Born charges and \(\epsilon _{\infty}\) was performed. The inputs and outputs can be found in the distribution.
The optimised results (KPOINTS, INCAR, POSCAR, CONTCAR, POTCAR) have been copied to the Standard_cell/Born/ directory making sure that CONTCAR has been copied to POSCAR. The KPOINTS file has been edited to increase the number of k-points to allow for the calculation of converged Born Charges.
The INCAR for a single point, with the calculation of epsilon infinity and Born charges(LEPSILON) is as follows:
PREC = accurate
ALGO = Normal
ADDGRID = T
NELMIN = 5
PREC = Accurate
IBRION = -1
NELMIN = 5
ENCUT = 500
EDIFF = 1.000000e-08
ISMEAR = 0
SIGMA = 1.000000e-02
IALGO = 38
LREAL = .FALSE.
LWAVE = .FALSE.
LCHARG = .FALSE.
LEPSILON = .TRUE.
LMAXMIX = 4
GGA = PS
The KPOINTS file is:
Regular k-point mesh
0
Gamma
6 6 2
0 0 0.5
Once the calculation is completed, the symmetry unique charges are calculated using the Phonopy script “phonopy-vasp-born”. Later on these will be converted to PDielec format, which has all of the atoms in the file, not just the symmetry unique ones.
phonopy-vasp-born > BORN
Standard Cell Phonopy calculation¶
From the Standard_cell/Born directory copy the BORN, POSCAR, POTCAR and KPOINTS files to Standard_cell/. The higher density of k-points is no longer needed, indeed the density may be reduced if a supercell is used.
Modify the INCAR file to do single point calculations with no storage of the charge density or the wavefunction:
PREC = Accurate
ALGO = Normal
IBRION = -1
NELMIN = 5
ENCUT = 500
EDIFF = 1.000000e-08
ISMEAR = 0
SIGMA = 1.000000e-02
LREAL = .FALSE.
LWAVE = .FALSE.
LCHARG = .FALSE.
LMAXMIX = 4
GGA = PS
ADDGRID = T
Since we will change the size of the cell, we can modify the KPOINTS file:
Regular k-point mesh
0
Monkhurst-Pack
2 2 1
0 0 0.5
phonopy -d --dim 2 2 1
mkdir SuperCell
cp SPOSCAR SuperCell/POSCAR
(cd SuperCell; cp ../INCAR .; ln -s ../POTCAR; ln -s ../KPOINTS)
Edit the SuperCell/INCAR file so that the wavefunction and charge are saved. The WAVECAR and CHGCAR files will restart the perturbed calculations. After running the VASP job in SuperCell/. Create links for the WAVECAR and CHGCAR files
ln -s SuperCell/WAVECAR; ln -s SuperCell/CHGCAR
Move the POSCAR-??? files for each displacement to a directory (POSCARS) where the calculations will be performed. POTCAR, KPOINTS and INCAR files are the same for every calculation, so link to the original SuperCell files. An advantage of using links for these files is that changes to the INCAR or KPOINTS files apply to all calculations. WAVECAR and CHGCAR links need to be made in each of the perturbed geometry directories.
mkdir POSCARS
for f in POSCAR-*
do
mkdir POSCARS/$f
cp $f POSCARS/$f/POSCAR
( cd POSCARS/$f; ln -s ../../INCAR; ln -s ../../POTCAR; ln -s ../../KPOINTS; ln -s ../../WAVECAR; ln -s ../../CHGCAR )
done
Submit all the jobs to a batch queue for processing.
cd POSCARS
for d in *
do
(cd $d; runvasp $d 8 8 )
done
Where runvasp is a script useds to create and run a job script to the batch queue
Calculate the FORCES_SETS¶
phonopy -f POSCARS/POSCAR-{001..005}/vasprun.xml
Calculate the Dynamical Matrix¶
The dynamical matrix is calculated in the standard basis, not the primitive.
phonopy --dim="2 2 1" --qpoints="0 0 0" --writedm
Calculate the full set of Born charges¶
PDielec needs the Born charges for every atom, not just the symmetry
unique ones. The full set of charges is generated in file “BORN_PDIELEC”
by the phonopy-pdielec-born command provided by PDielec. Note that
this command only uses the Phonopy API, no feature of the PDielec
API is used.
phonopy-pdielec-born > BORN_PDIELEC
For comparison the BORN file for provided in the distribution of Phonopy is;
# epsilon and Z* of atoms 1 13
3.27649624 -0.00000000 0.00000000 -0.00000000 3.27649624 0.00000000 0.00000000 0.00000000 3.21878866
2.96008813 -0.02948662 -0.00000000 0.02948662 2.96008813 -0.00000000 -0.00000000 0.00000000 2.92614183
-2.07328119 0.00000000 0.00000000 -0.00000000 -1.87350298 0.25811770 -0.00000000 0.35087130 -1.95076122
Whilst the BORN file calculated by the present calculation is;
# epsilon and Z* of atoms 1 13
3.27607426 -0.00000000 0.00000000 0.00000000 3.27607426 0.00000000 0.00000000 0.00000000 3.23683513
2.95996414 -0.02948839 -0.00000000 0.02948839 2.95996414 0.00000000 0.00000000 -0.00000000 2.92951772
-2.07319123 0.00000000 0.00000000 0.00000000 -1.87342763 0.25818161 0.00000000 0.35167360 -1.95301181
The agreement is accurate to 3 decimal places.
Standard Cell - PDGui¶
A pdgui calculation of the infrared intensities and the phonon frequencies is now possible.
pdgui phonopy.yaml
PDGui can be given any yaml file on the command line, the actual files read are always phonopy,yaml, qpoints.yaml and BORN_PDIELEC.
Primitive Cell Phonopy¶
A primitive cell calculation of the dynamical matrix will be performed in the Primitive_cell/ directory. Copy over files from the following files from the Standard_cell/ directory.
cd Standard_cell
cp BORN FORCE_SETS phonopy_disp.yaml phonopy.yaml ../Primitive_cell
phonopy --dim="2 2 1" --qpoints="0 0 0" --writedm --pa auto
phonopy-pdielec-born > BORN_PDIELEC
Comparison of results¶
In the following figures the results of the two different Phonopy calculations are compared. In addition results are shown for DFTP calculations on the same systems using VASP. The first figure shows a comparison over the full frequency range. The second figure compares the results over a limited frequency range to highlight the small differences between the calculations.

Fig. 67 Comparison of absorption predicted by Phonopy and DFTP (full frequencies)¶

Fig. 68 Comparison of absorption predicted by Phonopy and DFTP (restricted frequencies)¶
The results of the two DFPT calculations are indistinguishable from one another, as are those of the two Phonopy calculations. There is a small shift in the frequency peaks predicted by DFPT and Phonopy but this is to be expected.